What is progeria?
Hutchinson-Gilford Progeria Syndrome (HGPS) is a rare genetic disorder characterized by premature aging, caused by a point mutation in the LMNA gene.
This gene encodes many parts of the nuclear lamina: lamin A, lamin C, and lamin 10. Because of the affected gene, HGPS falls specifically under laminopathy, a certain class of conditions affecting the nuclear lamina, causing many changes to nuclear shape and genome stability.
In HGPS, the mutation creates a cryptic splice site in the LMNA gene, resulting in the continual production of progerin, a shortened and impaired protein. This changed protein is missing 50 amino acids compared to the normal lamin A and stays permanently farnesylated, thus leading to its toxic amassment into the nuclear lamina. Notably, lamin C along with lamin 10 are unaffected by that mutation.
How many people are affected?
According to the Progeria Research Foundation, it is estimated that between 350 and 400 children worldwide are currently living with this condition, making its prevalence around 1 in 20 million people.
What are the symptoms?
The destabilization of the nucleus, along with impaired genome integrity, is the primary cause of premature aging, the hallmark of HGPS. The resulting physiological alterations are widespread, affecting nearly all tissues.
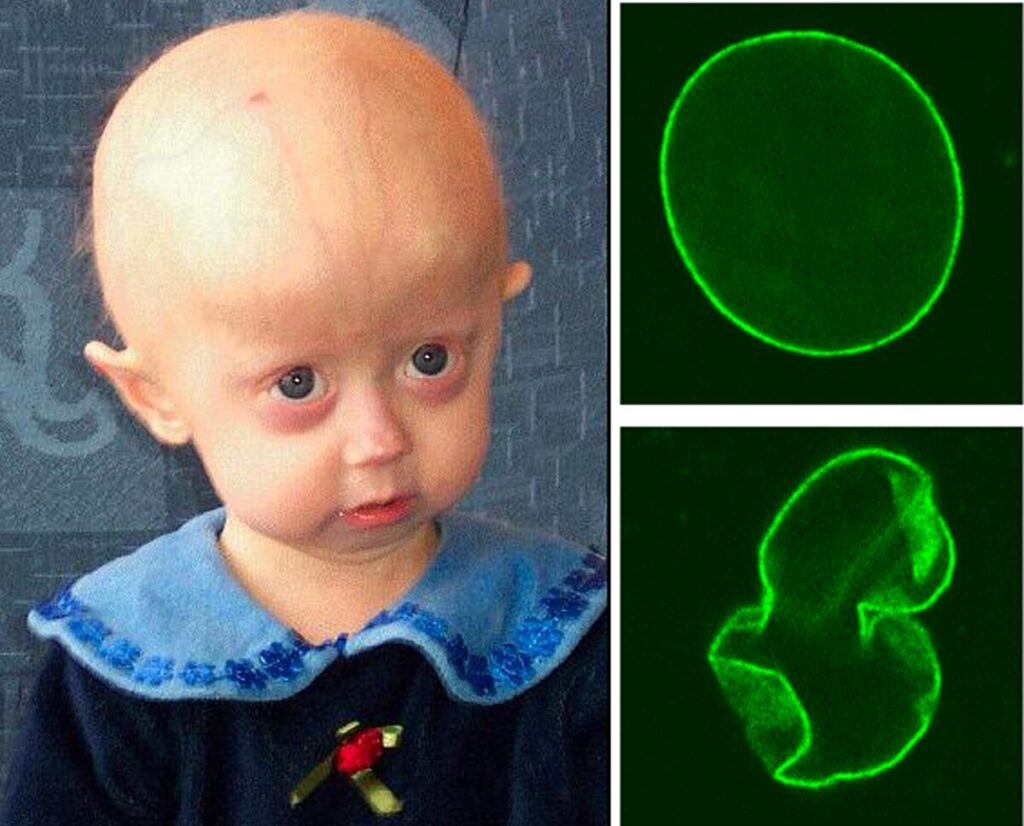
Children with HGPS exhibit distinctive features, including short stature, disproportionately small faces relative to their head size, prominent eyes, micrognathia (small jaw), and dental crowding. Additional symptoms include alopecia, loss of subcutaneous fat, fragile and thin skin, cardiovascular complications, and musculoskeletal abnormalities.
At the cellular level, progerin accumulation disrupts multiple biochemical pathways. Affected cells show increased levels of reactive oxygen species (ROS), aggregation of oxidized proteins, mitochondrial dysfunction, cellular senescence, and NF-κB activation, which leads to elevated pro-inflammatory cytokine levels.
First, progerin alters chromatin organization, leading to impaired transcriptional regulation and increased DNA damage, partly due to defects in repair mechanisms. The compromised nuclear architecture weakens the ability of cells to respond to genotoxic stress, contributing to genomic instability. Studies have shown that HGPS fibroblasts have an impaired DNA damage response, with reduced recruitment of repair proteins such as 53BP1 and Rad51. This defect accelerates cellular senescence and apoptosis, particularly in rapidly proliferating tissues, which could explain the severe phenotypic manifestations in fast-renewing tissues such as the epitelium. Additionally, studies have confirmed telomere shortening in fibroblasts derived from HGPS patients.
The nuclear rigidity caused by progerin accumulation affects mechanotransduction, the process by which the nucleus responds to mechanical signals from the extracellular environment, impairing cellular responses to mechanical stress. These alterations contribute to cellular senescence and systemic aging symptoms, particularly in tissues that undergo constant mechanical stress, such as the cardiovascular and musculoskeletal systems.
Another hallmark of HGPS cells is mitochondrial dysfunction, which leads to increased oxidative stress. Elevated levels of reactive oxygen species (ROS) further damage nuclear and mitochondrial DNA, creating a cycle of progressive cellular decline.
What is the life expectancy?
The leading causes of death in HGPS patients are complications related to atherosclerosis, various cardiac diseases, and strokes. Unfortunately, despite palliative treatments and emerging genetic engineering approaches, the current life expectancy for individuals with HGPS remains around 15 years.
What are the molecular pathways lending to the disease?
HGPS is an autosomal dominant disorder, meaning that a single mutant allele is sufficient to cause the disease. Progerin exerts a dominant-negative effect on normal prelamin A, interfering with the wild-type protein’s function.
The mutation that causes the disease is a C-to-T nucleotide substitution that does not alter the encoded amino acid sequence but creates a cryptic splice donor site. This misleads the spliceosome into producing a mature mRNA that is 150 nucleotides shorter, resulting in a truncated protein lacking 50 amino acids.
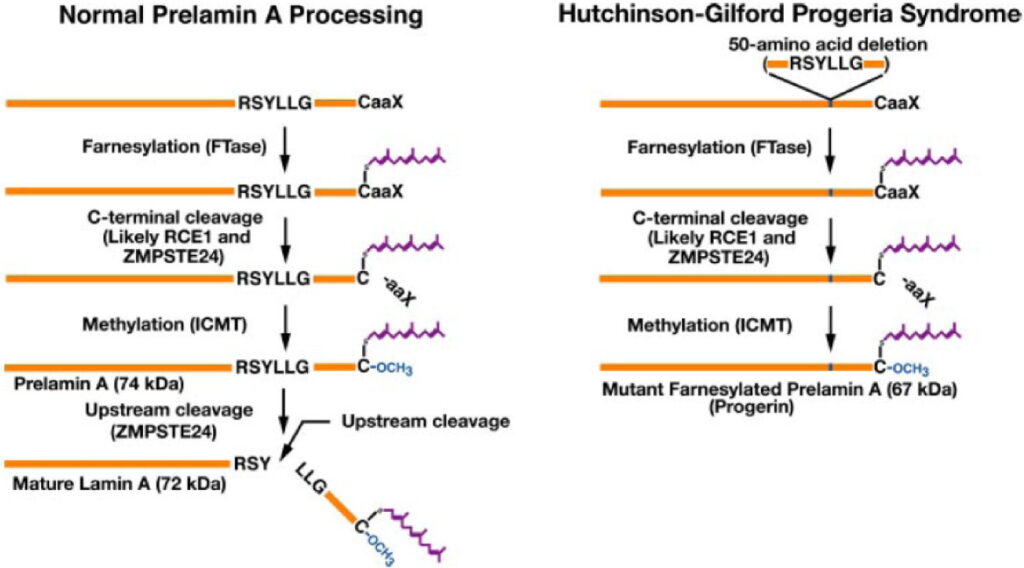
Physiologically, Lamin A is initially synthesized as a precursor called prelamin A, which undergoes a multi-step post-translational maturation process. This includes:
- Farnesylation of the C-terminal cysteine in its CaaX motif by farnesyltransferase.
- Cleavage of the last three amino acids by the metallopeptidase ZMPSTE24.
- Carboxymethylation of the remaining cysteine by isoprenylcysteine carboxyl methyltransferase.
- Final cleavage of the last 15 amino acids by ZMPSTE24, generating the mature, unfarnesylated lamin A.
The HGPS mutation activates a cryptic splice donor site in exon 11 of the LMNA gene, leading to the loss of the second ZMPSTE24 cleavage site. As a result, the truncated prelamin A (progerin) remains permanently farnesylated and accumulates abnormally in the nuclear lamina, causing multiple toxic effects.
Research gaps
Despite significant progress in understanding HGPS, several fundamental questions remain unanswered. One major challenge is determining why specific tissues are more severely affected than others. Given that progerin is expressed in all cells, it is unclear why certain tissues, such as the cardiovascular system and skin, exhibit the most pronounced pathological changes. The variability in disease severity among patients also suggests that unidentified genetic or epigenetic modifiers might influence disease progression.
Additionally, while progerin accumulation is clearly linked to cellular toxicity, its exact role in normal aging is still debated. Small amounts of progerin have been detected in healthy individuals, particularly in aged cells, suggesting that its accumulation might contribute to physiological aging. Understanding this connection could provide valuable insights into age-related diseases beyond HGPS.