Cos’è la HGPS?
La Sindrome di Hutchinson-Gilford (HGPS) è una malattia monogenica estremamente rara, caratterizzata da un invecchiamento precoce. È causata da una mutazione puntiforme de novo nel gene LMNA, che codifica tre componenti della lamina nucleare: lamin A, lamin C e lamin 10. Poiché la mutazione colpisce questo gene, l’HGPS è classificata come una laminopatia, un gruppo di patologie che alterano la struttura della lamina nucleare, compromettendo la morfologia del nucleo e l’integrità del genoma.
L’HGPS segue un modello di ereditarietà autosomica dominante, il che significa che la presenza di un singolo allele mutato è sufficiente a causare la malattia. La proteina anomala prodotta, progerina, esercita un effetto dominante negativo sulla prelamina A normale, interferendo con la funzione della proteina wild-type. È importante sottolineare che lamin C e lamin 10 non subiscono alterazioni a causa di questa mutazione.
Quante persone ne sono colpite?
Secondo la Progeria Research Foundation, si stima che attualmente tra 350 e 400 bambini nel mondo convivano con questa condizione, con una prevalenza di circa 1 caso ogni 20 milioni di persone.
Quali sono i sintomi?
L’instabilità nucleare e il danneggiamento del genoma sono i principali fattori che portano all’invecchiamento precoce, la caratteristica distintiva dell’HGPS. Le alterazioni fisiologiche risultanti sono diffuse e colpiscono quasi tutti i tessuti.
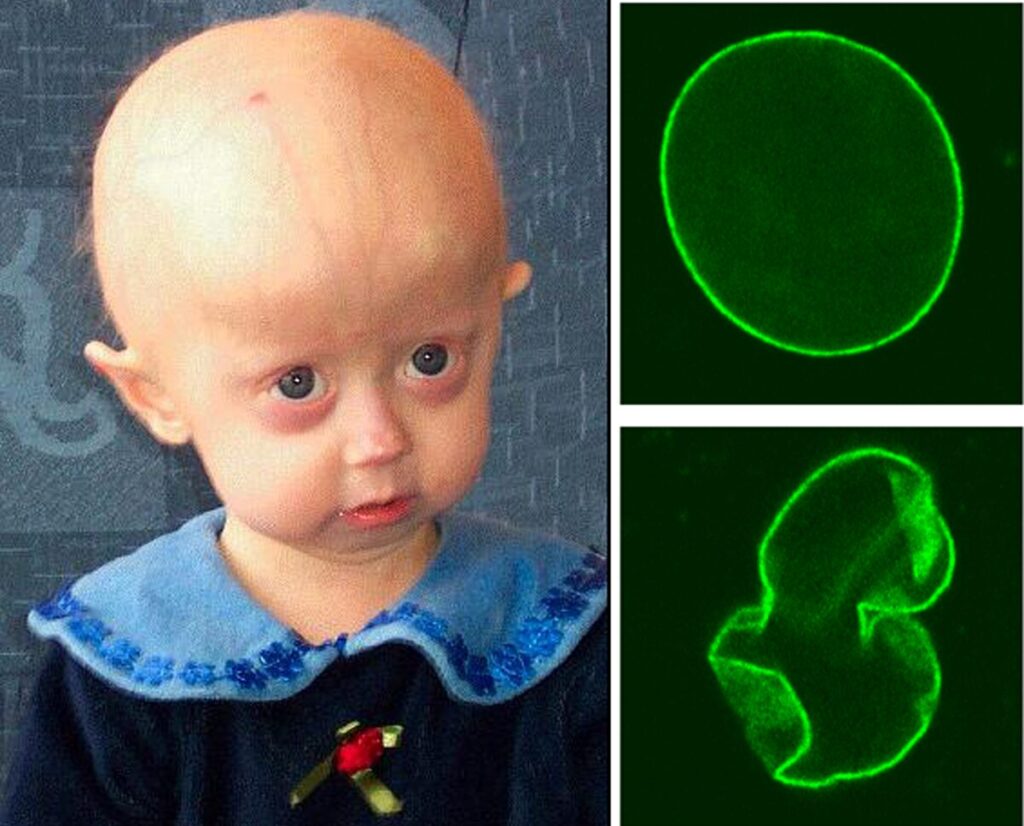
I bambini affetti da HGPS presentano tratti distintivi come bassa statura, un viso sproporzionatamente piccolo rispetto alla testa, occhi prominenti, micrognazia (mascella piccola) e affollamento dentale. Altri sintomi includono alopecia, perdita del grasso sottocutaneo, pelle sottile e fragile, complicanze cardiovascolari e anomalie muscoloscheletriche.
A livello cellulare, l’accumulo di progerina altera molteplici vie biochimiche. Le cellule affette mostrano un aumento dei livelli di specie reattive dell’ossigeno (ROS), aggregazione di proteine ossidate, disfunzione mitocondriale, senescenza cellulare e attivazione di NF-κB, con conseguente incremento della produzione di citochine pro-infiammatorie.
Oltre all’instabilità nucleare, la progerina compromette processi cellulari essenziali. In particolare, altera l’organizzazione della cromatina, portando a una regolazione trascrizionale difettosa e a un aumento del danno al DNA, in parte dovuto a difetti nei meccanismi di riparazione.
L’architettura nucleare compromessa riduce la capacità delle cellule di rispondere allo stress genotossico, contribuendo all’instabilità genomica. Studi hanno dimostrato che i fibroblasti di pazienti con HGPS presentano un difetto nella risposta al danno del DNA, con un reclutamento ridotto di proteine di riparazione come 53BP1 e Rad51. Questo deficit accelera la senescenza cellulare e l’apoptosi, in particolare nei tessuti a rapido turnover, il che potrebbe spiegare le gravi manifestazioni fenotipiche nei tessuti a elevato rinnovo, come l’epitelio. Inoltre, studi hanno confermato un accorciamento dei telomeri nei fibroblasti derivati da pazienti con HGPS.
La rigidità nucleare causata dall’accumulo di progerina influenza la meccanotrasduzione, ovvero il processo attraverso cui il nucleo risponde ai segnali meccanici provenienti dall’ambiente extracellulare, compromettendo la capacità delle cellule di rispondere allo stress meccanico. Queste alterazioni contribuiscono alla senescenza cellulare e ai sintomi sistemici dell’invecchiamento, in particolare nei tessuti sottoposti a costante sollecitazione meccanica, come il sistema cardiovascolare e quello muscoloscheletrico.
Un altro aspetto distintivo delle cellule HGPS è la disfunzione mitocondriale, che porta a un aumento dello stress ossidativo. L’elevata produzione di specie reattive dell’ossigeno (ROS) causa ulteriori danni al DNA nucleare e mitocondriale, innescando un ciclo di progressivo declino cellulare. Complessivamente, questi meccanismi mostrano come la tossicità della progerina vada oltre le anomalie nucleari, influenzando numerose funzioni cellulari essenziali.
Qual è l’aspettativa di vita?
Le principali cause di morte nei pazienti con HGPS sono le complicanze dell’aterosclerosi, le patologie cardiache e ictus. Purtroppo, nonostante i trattamenti palliativi e le nuove tecniche di ingegneria genetica, l’aspettativa di vita attuale per le persone affette da questa malattia rimane intorno ai 15 anni.
Quali sono le vie molecolari che portano alla sindrome?
La mutazione responsabile è una sostituzione nucleotidica C>T nell’esone 11 del gene LMNA del che si trova sul cromosoma 1. La sostituzione è silente, ovvero non altera la sequenza amminoacidica codificata, ma crea tuttavia un sito donatore di splicing criptico. Questo induce lo spliceosoma a produrre un mRNA maturo più corto di 150 nucleotidi, con una conseguente proteina tronca priva di 50 amminoacidi.
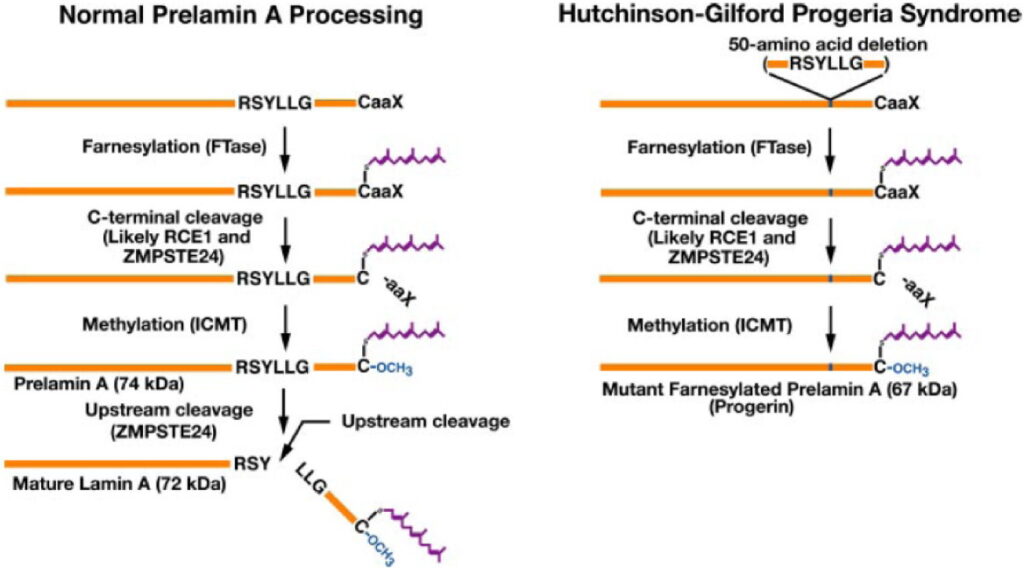
A livello fisiologico, la lamina A viene inizialmente sintetizzata come precursore, chiamato prelamina A, e matura attraverso un processo post-traduzionale a più stadi, che include:
- Farnesilazione della cisteina terminale nel motivo CaaX da parte della farnesiltransferasi.
- Scissione degli ultimi tre amminoacidi da parte della metalloproteasi ZMPSTE24.
- Carbossimetilazione della cisteina rimanente da parte dell’enzima isoprenilcisteina carbossimetiltransferasi.
- Scissione finale degli ultimi 15 amminoacidi da parte della ZMPSTE24, generando la lamina A matura e defarnesilata.
La mutazione patologica che comporta HGPS attiva un sito donatore di splicing criptico nell’esone 11 del gene LMNA, impedendo la seconda scissione mediata da ZMPSTE24. Di conseguenza, la prelamina A tronca (progerina) rimane permanentemente farnesilata e si accumula in modo anomalo nella lamina nucleare, esercitando effetti tossici multipli.
Lacune nella Ricerca
Nonostante i significativi progressi nella comprensione dell’HGPS, restano ancora diverse domande senza risposta. Una delle principali sfide è determinare perché alcuni tessuti siano più gravemente colpiti rispetto ad altri. Dato che la progerina è espressa in tutte le cellule, non è ancora chiaro il motivo per cui certi tessuti, come il sistema cardiovascolare e la pelle, manifestino le alterazioni patologiche più pronunciate. Inoltre, la variabilità nella gravità della malattia tra i pazienti suggerisce che possano esistere modificatori genetici o epigenetici non ancora identificati che influenzano la progressione della patologia.
Inoltre, sebbene l’accumulo di progerina sia chiaramente legato alla tossicità cellulare, il suo ruolo esatto nell’invecchiamento normale è ancora oggetto di dibattito. Piccole quantità di progerina sono state rilevate anche in individui sani, in particolare nelle cellule invecchiate, suggerendo che il suo accumulo possa contribuire all’invecchiamento fisiologico. Comprendere questa connessione potrebbe fornire preziose informazioni sulle malattie legate all’età oltre all’HGPS.